Występują raz na milion urodzeń, pojawiają się w dzieciństwie, a na dodatek zazwyczaj kończą źle. Mowa o chorobach rzadkich. Oto 10 takich schorzeń, które występują ekstremalnie rzadko.
Większość z nich to choroby genetyczne, które ujawniają się zaraz po urodzeniu lub we wczesnym dzieciństwie. Niestety, rokowania w przypadku ich wystąpienia nie są zazwyczaj dobre. Mimo ogromnych postępów w medycynie, skutecznych leków na nie brak. Problem ten dotyczy ok. 7-8% populacji, czyli aż 350 mln ludzi na świecie.
Syndrom MDP
To prawdopodobnie najrzadsza choroba na świecie. Syndrom MDP (ang. Mandibular hypoplasia, Deafness, Progeroid features) zdiagnozowano do tej pory u 8 osób. Nie magazynują one tkanki tłuszczowej, mimo że ich organizm prawidłowo wchłania wszystkie inne składniki odżywcze. Choroba ma podłoże genetyczne. Nie ma na nią żadnego lekarstwa, chociaż naukowcy eksperymentują z techniką edycji genów CRISPR/Cas9.
AHC
Inną rzadką chorobą jest AHC (Alternating Hemiplegia of Childhood), czyli naprzemienna hemiplegia dziecięca. Trafia się około raz na milion urodzeń. Jest trudna do zdiagnozowania, bo większość lekarzy o niej zwyczajnie nie słyszała. U większości chorych błędnie diagnozuje się lekooporną padaczkę lub mózgowe porażenie dziecięce. Choroba objawia się epizodami porażenia połowiczego – od lekkiego do całkowitego. Wygląda to tak, jakby ciało wykrzywiała jakaś niewidzialna siła. W przeszłości była mylona z opętaniami przez demony.
Fibrodysplazja

Fibrodysplazja, czyli postępujące kostniejące zapalenie mięśni (FOP) to rzadka choroba genetyczna, która objawia się stopniowym kostnieniem tkanek miękkich. Częstość jej występowania to 0,6 przypadków na milion. Choroba jest powodowana przez mutację genu ACVR1. Dochodzi do nadekspresji białka BMP4, które stymuluje tworzenie naczyń krwionośnych, rozrost tkanki łącznej i wytwarzanie kości. Doprowadza to do postępującego skostnienia tkanki mięśniowej i tkanki łącznej. Na świecie jest ok. 800 chorych. Nie jest znane skuteczne leczenie.
Syndrom Booka
Syndrom Booka to bardzo rzadka choroba, na której obraz kliniczny składa się aplazja zębów przedtrzonowych, nadmierna potliwość i przedwczesne siwienie. Do tej pory w literaturze opisano jedynie 25 przypadków zachorowań. Co ciekawe, wszystkie odnotowane chore osoby były ze sobą spokrewnione w obrębie czterech pokoleń. Dodatkowe zaobserwowane objawy to wąskie podniebienie, słabo wykształcone paznokcie, małe dłonie i anomalie dotyczące brwi i linii papilarnych.
Kuru

Kuru, czyli „śmiejąca się śmierć”, jest śmiertelną, nieuleczalną chorobą zakaźną wywoływaną przez priony. Jest to pierwsza opisana zakaźna encefalopatia gąbczasta u ludzi. Do zakażenia kuru dochodziło u praktykujących kanibalizm członków plemienia Fore, zamieszkującego Papuę-Nową Gwineę. W trakcie przebiegu choroby, zawsze pojawia się ataksja móżdżkowa, rzadko zaś nie występuje otępienie (w przeciwieństwie do innych encefalopatii wywoływanych przez priony). U chorych pojawiają się prymitywne odruchy (np. ssania, gryzienia), a także huśtawki nastrojów – od depresji po euforię. Podobnie jak w innych chorobach prionowych, nie jest stosowane żadne leczenie przyczynowe.
Dysplazja Lewandowsky’ego-Lutza

Dysplazja Lewandowsky’ego-Lutza (EV) to rzadka choroba skóry o podłożu genetycznym. Uważa się, że jest ona wywołana przez wirusy HPV u osób z mutacją w obrębie jednego z dwóch sąsiadujących genów: EVER1 lub EVER2. Osoby chore często są nazywane ludźmi-drzewami. Wykwity w EV pojawiają się już w dzieciństwie – mają postać płaskich grudek, plam rumieniowych lub przebarwień. Stanowią one punkt wyjścia dla raków skóry. Leczenie retinoidami jest nieskuteczne i daje jedynie przejściową poprawę. Zmiany nowotworowe usuwa się chirurgicznie.
Progeria
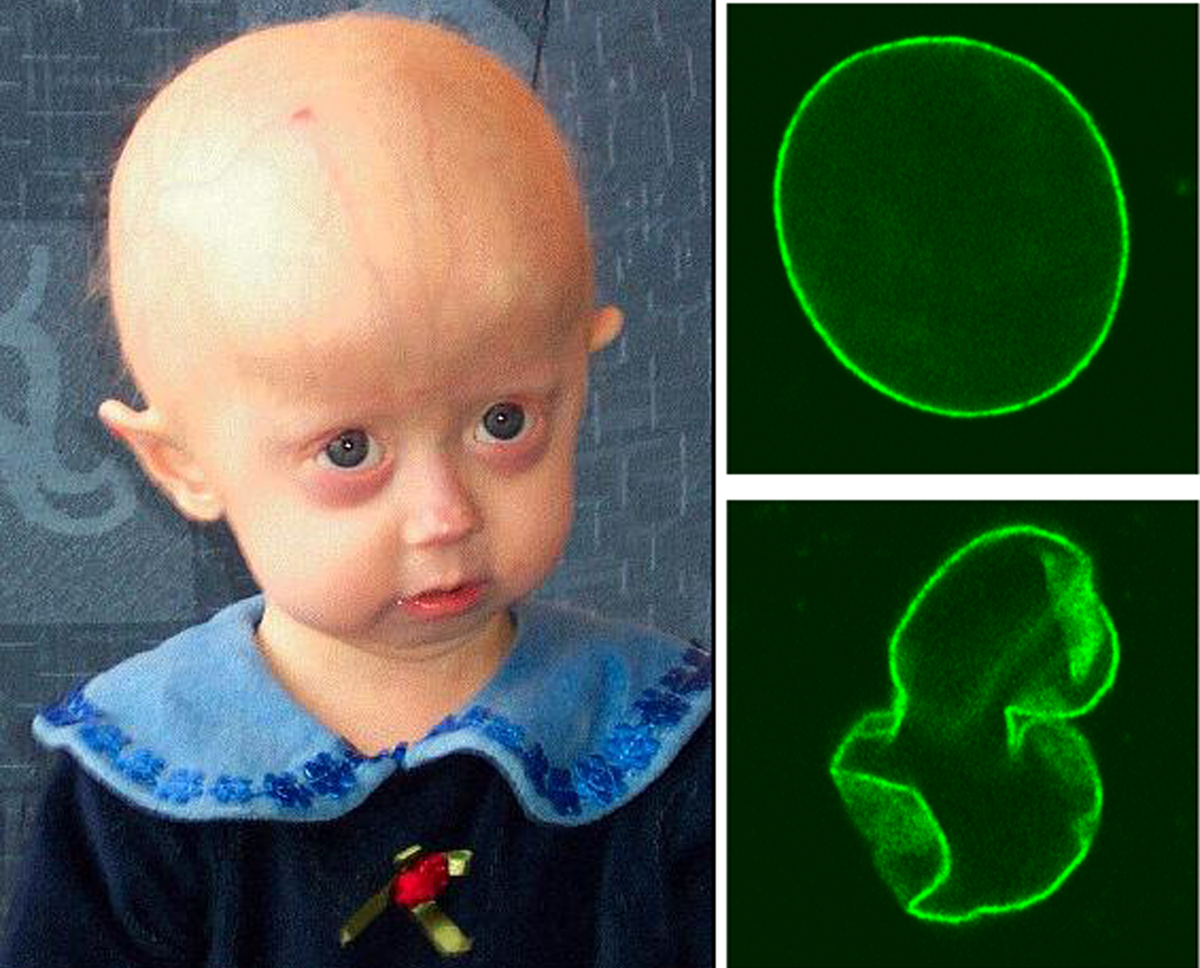
Progeria to rzadka choroba genetyczna, która charakteryzuje się przyspieszonym procesem starzenia. Jest wywołana mutacją w genie LMNA kodującym laminę A. Białko to stabilizuje błonę otaczającą jądro komórkowe, a jego mutacje powodują zaburzenia w strukturze i funkcjach jądra. Średnia długość życia osób cierpiących na progerię to 13 lat. Nie istnieje żadna metoda leczenia przyczynowego progerii, nie ma także żadnych zaleceń dla pacjentów z tą chorobą. Zgon występuje najczęściej z powodu chorób układu krążenia.
Zespół Alstroma
Zespół Alstroma (ALMS) jest rzadką chorobą genetyczną, która objawia się cukrzycą typu 2, retinopatią barwnikową, głuchotą, otyłością i hipogonadyzmem. Do tej pory odnotowano 900 przypadków zachorowań na całym świecie. Jej przyczyną jest mutacja genu ALMS1, którą dziedziczy się od obojga rodziców. Gen ten koduje białko o nieznanej funkcji – nie wiadomo, co dokładnie robi w organizmie. Niektóre osoby cierpiące na ALMS dotyka także nietypowe schorzenie skóry, przejawiające się jej nienaturalną grubością, aksamitną fakturą i ciemnym zabarwieniem.
Zespół Seckela
Zespół Seckela to rzadki zespół wad wrodzonych, które cechuje wewnątrzmaciczne i pourodzeniowe zahamowanie wzrostu, upośledzenie umysłowe i dysmorfia twarzy. Zidentyfikowano co najmniej trzy geny, które są odpowiedzialne za rozwój choroby: SCKL1, SCKL2 i SCKL3. Osoby chore cechuje niski wzrost oraz charakterystyczne zniekształcenie twarzy (mikrocefalia, duże oczy, dziobowaty nos). Zmianom tym zwykle towarzyszy ciężkie opóźnienie umysłowe. Do tej pory odnotowano niewiele ponad 100 przypadków.
Płód arlekin
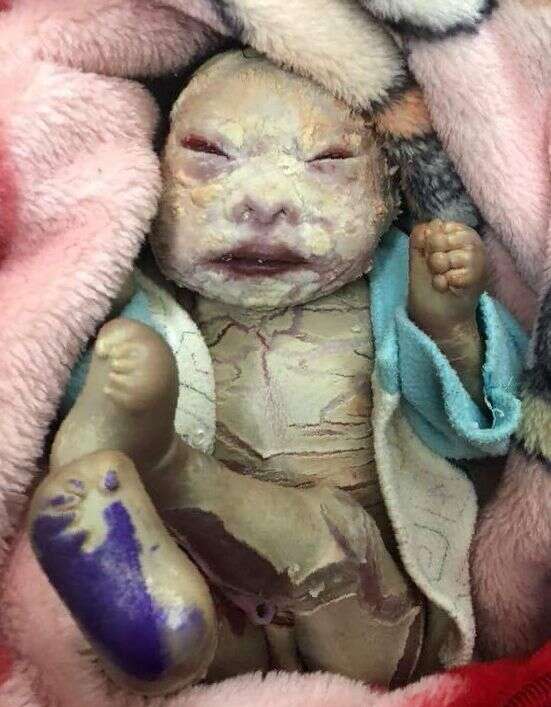
Płód arlekin to wyjątkowo rzadka choroba genetyczna, która należy do genodermatoz. Charakteryzuje się pokrywającymi całą powierzchnię ciała łuskami hiperkeratotycznego naskórka o kształcie wielokątów. Ich ułożenie przywodzi na myśl kostium arlekina. Poza tym stwierdza się niską masę urodzeniową, erytrodemię, wywinięcie warg i powiek. Przynajmniej część przypadków wiąże się z mutacjami genu ABCA12. W literaturze opisano do tej pory ponad 100 przypadków choroby. Częstość jest wyższa w zamkniętych społecznościach, gdzie dochodzi do aktów kazirodczych, np. wśród Indian Navajo.