Proces tworzenia nowych leków jest skomplikowany i czasochłonny. Jednym z powodów tego stanu rzeczy jest samo poszukiwanie odpowiednich cząsteczek leków, które są w stanie wiązać się z odpowiednimi białkami w naszym organizmie. Coraz częściej jednak do tego zadania zespoły badawcze korzystają ze sztucznej inteligencji.
I tak samo jest w przypadku systemu GeoMol, stworzonego przez zespół naukowców z MIT. GeoMol to algorytm maszynowego uczenia się, który na podstawie dwuwymiarowego schematu danej cząsteczki jest w stanie zaprezentować jej trójwymiarowy model, dzięki czemu jesteśmy w stanie zobaczyć w jaki sposób jest ona w stanie przyłączyć się do określonych białek w naszym organizmie.
GeoMol tworzy trójwymiarowe modele analizowanych cząsteczek w zaledwie kilka sekund i działa lepiej niż inne modele uczenia maszynowego wykorzystywane do tego typu badań. Twórcy algorytmu twierdzą, że ich dzieło może pomóc firmom farmaceutycznym w procesie tworzenia nowych leków, znacznie go przyspieszając dzięki zawężeniu liczby potencjalnych cząsteczek, które muszą zostać przetestowane w eksperymentach laboratoryjnych.
Jak działa trójwymiarowe modelowanie cząsteczek?
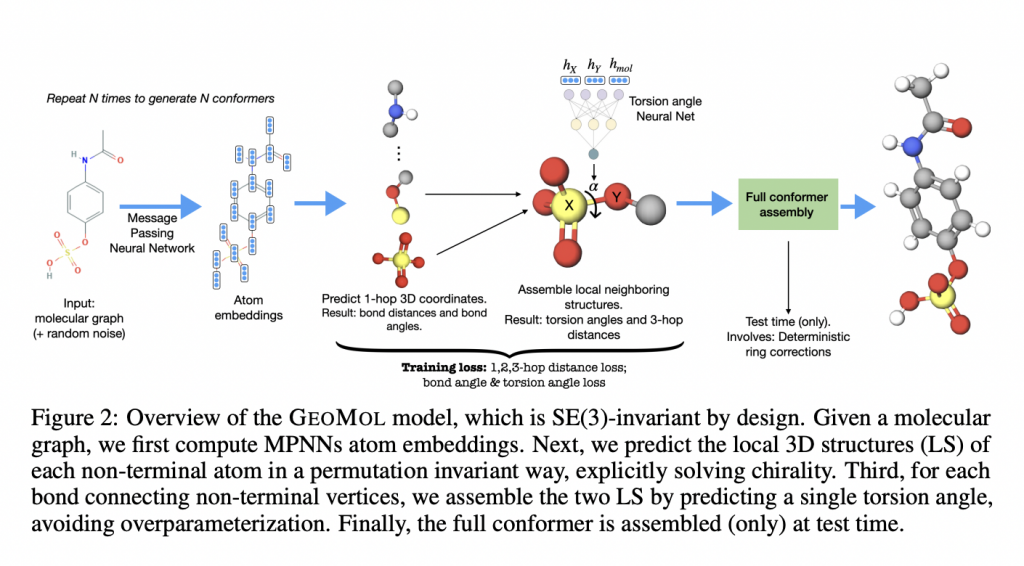
Otrzymując wykres molekularny jako daną wejściową, GeoMol wstępnie przewiduje długości wiązań chemicznych między poszczególnymi atomami oraz kąty pojedynczych wiązań między nimi. Sposób ułożenia i połączenia atomów określa, które wiązania mogą się obracać. Następne w kolejce jest przewidywanie indywidualnej struktury lokalnego sąsiedztwa dla każdego atomu – algorytm prognozuje jak wyglądać będą sąsiednie pary wiązań obrotowych i oblicza ich kąty skręcania.
— Wiązania obrotowe mogą przyjmować szeroki zakres możliwych wartości. Tak więc użycie naszego algorytmu pozwala nam uchwycić wiele lokalnych i globalnych środowisk, które wpływają na to przewidywanie. Obrotowe wiązania mogą przyjmować wiele wartości i chcemy, aby nasze przewidywania odzwierciedlały ten podstawowy rozkład – mówi Lagnajit Pattanaik, doktorant na Wydziale Inżynierii Chemicznej i współtwórca GeoMola.
GeoMol nie osiągnął jeszcze pełni swoich możliwości
Kolejną, ogromnie ważną zaletą algorytmu jest to, że potrafi on przewidywać chiralność analizowanej cząsteczki, co skutkuje dodatkowo zaoszczędzonym czasem, jeśli chodzi o badania laboratoryjne. W przyszłości naukowcy mają nadzieję zastosować algorytm GeoMol w obszarze wysokoprzepustowych wirtualnych badań przesiewowych, wykorzystując model do określenia struktur małych cząsteczek, które będą oddziaływać z określonym białkiem.
Czytaj również: Komputer i projektowanie leków? Alphabet powołał do życia nową firmę
W międzyczasie GeoMol będzie oczywiście udoskonalany za pomocą kolejnych danych treningowych, dzięki którym jego możliwości przewidywania trójwymiarowych struktur bardziej skomplikowanych cząsteczek powinny wzrosnąć.
Największą zaletą wykorzystania tego typu algorytmów jest ogromna oszczędność czasu, jeśli chodzi o poszukiwanie i projektowanie nowych leków. W przyszłości zapewne proces ten uda się zautomatyzować jeszcze bardziej – tj. algorytmy, oprócz analizy i przewidywania struktur, będą potrafiły również samodzielnie proponować cząsteczki z najwyższym potencjałem, jeśli chodzi o tworzenie nowych leków.